SMUG: Somatic MUtation Gleaner
URL of this page: http://biostat.mc.vanderbilt.edu/SMUG Program: SMUG.tgz. To uncompress, typetar zxf SMUG.tgz.
Introduction
Somatic Mutation Gleaner (SMUG) was developed to effectively detect base substitutions and loss of heterozygosity (LOH) using next-generation sequencing data for normal and tumor tissues. It first screens bam files using walker programs (modules we developed to run under GATK), and then summarizes the results using Perl scripts. Here is a brief description of the algorithms used by SMUG.Detection of base substitutions: For each patient, we screen the genome for sites that are homozygous in the normal sample and have variants in the tumor sample. To adjust for depth variation, an empirical Bayes (EB) estimate of tumor variant rate is calculated. To minimize the effect of sequencing/alignment errors, the tumor EB-adjusted rate is further adjusted using the normal sample variant rate as baseline. (A site may be called homozygous with a high GQ in the normal sample and heterozygous with a high GQ in the tumor sample when both samples appear to carry similar fractions of variant alleles.)
The results will be consolidated across patients. The user can control how much to output. For example, the user may choose to output sites with mutants in at least two patients. Please see below for details.
Detection of LOH: For each patient, we screen the genome for sites that are heterozygous in the normal sample and have a significant departure from 50:50 in allele counts in the tumor sample. An LOH score is calculated for such a site. An LOH region is defined to be a region containing contiguous sites with high LOH scores. The user can specify the criteria for defining an LOH region. Please see below for details.
Paper
Song Z, Long J, He J, Shi J, Shu XO, Cai Q, Zheng W, Li C (2012) Efficient detection of tumor somatic mutations using next-generation sequencing data. (to be submitted)Contact
Chun Li: chun.li@vanderbilt.edu Zhuo Song: zhuo.song@vanderbilt.eduI. Installation
Please follow the steps blow to download and build GATK and SMUG.
- Make sure you have installed JDK, Ant, and Git.
- Create a GATK folder and download GATK source from its Git repository.
mkdir GATK git clone git://github.com/broadgsa/gatk.git GATK
- Download SMUG source.
wget http://biostat.mc.vanderbilt.edu/wiki/pub/Main/SMUG/SMUG.tgz tar xzf SMUG.tgz
- Copy the SMUG_walkers folder to the GATK walker folder.
cp -r SMUG/SMUGwalkers/ GATK/public/java/src/org/broadinstitute/sting/gatk/walkers/
- Build the SMUG walkers and GATK from source code. SMUG has been tested on GATK version 1.6-2-gc2b74ec (05/02/2012).
cd GATK git checkout 1.6-2-gc2b74ec ant clean ant
git checkout command ensures the posted version of SMUG can run successfully. To update GATK if you already have a clone of GATK:
cd GATK git pullIf you experience problems running GATK after updating via
git pull,
try cleaning your clone and recompiling GATK following step 5 above.
II. Running SMUG walkers
The SMUG program contains three GATK walkers, two Perl scripts, and an R program. The Perl scripts and R program will be explained in sections III and IV. The walkers are:- SMUG_BaseSub: for detection of base substitutions
- SMUG_LOH: for detection of LOH
- SMUG: for detection of both base substitutions and LOH. If the user wants both detections, the SMUG walker is recommended as it will save the total running time.
NOTE: A walker needs to be run separately for each patient.
- To display help messages for the walkers:
java -jar GATK/dist/GenomeAnalysisTK.jar -T SMUG -h java -jar GATK/dist/GenomeAnalysisTK.jar -T SMUG_BaseSub -h java -jar GATK/dist/GenomeAnalysisTK.jar -T SMUG_LOH -h
Options
-R,--reference_sequence reference sequence file
-I,--input_file list of SAM or BAM files (see below)
-D,--dbsnp dbSNP file; optional but recommended
-o,--out log file; default to screen
-smT,--SampleName_Tumor sample ID of tumor tissue; it is the SN
tag in sam/bam files
-smN,--SampleName_Normal sample ID of normal tissue; it is the SN
tag in sam/bam files
-typeT,--SampleType_Tumor sample type of tumor tissue, user defined
name for tumor sample, default = 'Tumor'
-typeN,--SampleType_Normal sample type of normal tissue, user defined
name for normal sample, default = 'Normal'
-minBQ,--minBaseQ minimum base quality, default = 20
-minMQ,--minMappingQ minimum mapping quality, default = 20
-minAllN,--minAllAlleles_Normal minimum base count in Normal sample after
filtering by BQ and MQ, default = 10
-minAllT,--minAllAlleles_Tumor minimum base count in Tumor sample after
filtering by BQ and MQ, default = 5
-minRestT,--minMinorAlleles_Tumor minimum total base count for the minor
alleles in Tumor sample after filtering
by BQ and MQ, default = 2;
Not applicable to the SMUG_LOH walker
-minHRT,--minHeteroRate_Tumor minimun fraction of minor alleles in Tumor
sample (RestT/AllT), default = 0.1;
Not applicable to the SMUG_LOH walker
-stand_call_conf minimum confidence for genotype calling
(genotyping quality, GQ), default = 30
In addition, some arguments of GATK UnifiedGenotypers can also be specified (please
consult GATK documentations for details). For example, if the user
wants to run our walkers only on a few regions, the -L option can be
used:
-L,--intervals <intervals>Detailed explanations of the options:
- All options except
-Dare required, but most of them have default values if unspecified. Although-Dis optional, it is highly recommended. - The file for the
-Ioption contains the path to bam/sam files, one file per line. The order of files is not important as long as there are data matching normal and tumor sample IDs specified by the options-smNand-smT. - Once GATK finishes pileup of all reads from bam files, it will stratify reads based on the SN tag (sample ID). Then SMUG calls the following built-in function to process the data:
AlignmentContextUtils.splitContextBySampleName(context, sampleID) - The options
-typeNand-typeTare defined by the user, the default values are "Normal" and "Tumor". They can be any string without space to help the user to describe the data. They will be used as part of the output file names to easy identification of files.
Example command
java -jar GATK/dist/GenomeAnalysisTK.jar -T SMUG \ -R human_b36_male.fa \ -D:VCF dbsnp_132.b36.vcf \ -I normal_and_tumor_samples_bam.list \ -o SMUG.txt \ -smN normal_sample_ID \ -smT tumor_sample_ID \ -typeN Normal \ -typeT Tumor \ -minBQ 20 \ -minMQ 20 \ -minAllN 10 \ -minAllT 10 \ -minRestT 2 \ -minHRT 0.01
Log file
The log file specified through-o contains the information on
parameter values, result files, and number of loci reported. If the
user does not specify -o, the information will be printed to the
screen. Here is an example log file:
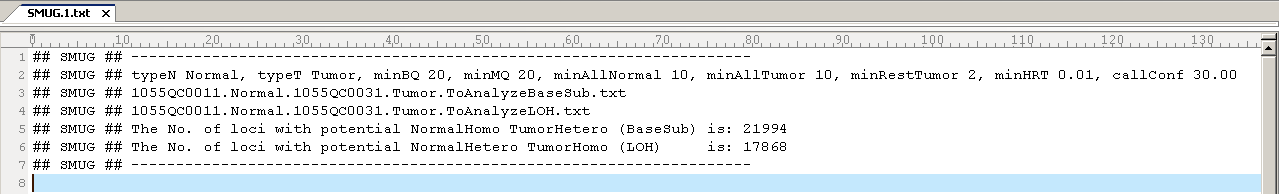
Output of the SMUG_BaseSub walker
The SMUG_BaseSub walker will generate result files for further analysis by the SMUG_BaseSub.pl script (see section III). The file names will follow the following pattern:
- "smN"."typeN"."smT"."typeT".ToAnalyzeBaseSub.txt
Here is an example result file of the SMUG_BaseSub walker:
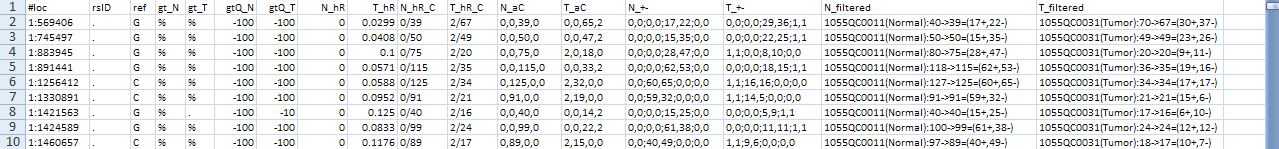 It has 17 columns:
It has 17 columns:
loc chr:position
rsID rs# or '.'
ref reference allele
gt_N normal genotype: '.' no call, '%' no variation, 'N/N'
(N=A,C,G,T), * means ref
gt_T tumor genotype
gtQ_N normal genotype quality, -10 means '.', -100 means '%'
gtQ_T tumor genotype quality, -10 means '.', -100 means '%'
N_hR normal variant rate
T_hR tumor variant rate
N_hR_C normal variant rate in 'variant count/all allele count' format
T_hR_C tumor variant rate in 'variant count/all allele count' format
N_aC normal sample base counts: a;c;g;t
T_aC tumor sample base counts: a;c;g;t
N_+- normal sample base counts by strand: a+,a-;c+,c-;g+,g-;t+,t-
T_+- tumor sample base counts by strand: a+,a-;c+,c-;g+,g-;t+,t-
N_filtered normal sample read counts with format
sampleID(sampleType):rawReads->fillteredReads=(reads+,reads-);
for example: 5QC011(Normal):94->92=(57+,35-), meaning
sample '5QC011' has 94 reads covering the site, 92
passing MQ/BQ filtering (57 on '+', 35 on '-' strand)
T_filtered tumor sample read counts
Note that some columns may have information overlap.
Output of the SMUG_LOH walker
The SMUG_LOH walker will generate result files for further analysis by the SMUG_LOH.pl script (see section IV). The file names will follow the following pattern:
- "smN"."typeN"."smT"."typeT".ToAnalyzeLOH.txt
Here is an example result file of the SMUG_LOH walker:
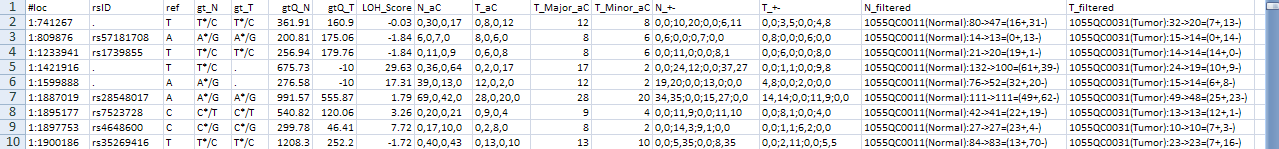
It has 16 columns:
loc chr:position
rsID rs# or '.'
ref reference allele
gt_N normal genotype: '.' no call, '%' no variation, 'N/N'
(N=A,C,G,T), * means ref
gt_T tumor genotype
gtQ_N normal genotype quality, -10 means '.', -100 means '%'
gtQ_T tumor genotype quality, -10 means '.', -100 means '%'
LOH_Score LOH score for this site
N_aC normal sample base counts: a;c;g;t
T_aC tumor sample base counts: a;c;g;t
T_Major_aC major allele counts in tumor sample
T_Minor_aC minor allele counts in tumor sample
N_+- normal sample base counts by strand: a+,a-;c+,c-;g+,g-;t+,t-
T_+- tumor sample base counts by strand: a+,a-;c+,c-;g+,g-;t+,t-
N_filtered normal sample read counts with format
sampleID(sampleType):rawReads->fillteredReads=(reads+,reads-);
for example: 5QC011(Normal):94->92=(57+,35-), meaning
sample '5QC011' has 94 reads covering the site, 92
passing MQ/BQ filtering (57 on '+', 35 on '-' strand)
T_filtered tumor sample read counts
III. Running SMUG_BaseSub.pl
The script does the following:- Read in the SMUG_BaseSub output files (*.ToAnalyzeBaseSub.txt).
- For each patient, keep the sites qualifying the criteria specified by the user.
- For each patient, calculate the empirical Bayes (EB) estimates (alpha and beta) using selected sites in tumor data, and apply the EB-adjustment to both tumor and normal tissue data. (This step is performed using the R program SMUG_BaseSub.EB.R).
- Summarize statistics for all sites cross all patients, and report them. The sites are ordered by the 'sumDiff' statistic (sum across patients of the differences between EB-adjusted tumor and normal rates (see section V for details on file format). As the result file is a text file, the user can easily sort the sites using other criteria.
Arguments for SMUG_BaseSub.pl
-L file containing names of SMUG_BaseSub output files, one on each line
-restEB minimum number of alternative alleles for EB parameter estimation,
default = 3
-allEB minimum depth for EB parameter estimation, default = 10
-nVote minimum number of patients carrying base substitution, default = 2
-restT minimum number of alternative alleles in Tumor for reporting,
default = 2
-allT minimum depth in Tumor for reporting, default = 10
-maxT_hR maximum tumor variant rate, default = 0.9
The -maxT_hR option is for filtering out sites with very high tumor variants
rate. These are likely due to alignment artifacts rather than real
mutations.
The -restEB and -allEB options are for estimating the EB-parameters
alpha and beta. We have found the default values work well for our data.
Example command to run SMUG_BaseSub.pl
perl SMUG_BaseSub.pl -L BaseSub.list \ -restEB 3 -allEB 10 \ -nVote 2 \ -restT 2 -allT 10
Output files of SMUG_BaseSub.pl
There is a main output file (one file for all patients) with file name following the pattern:
- "L".nVote."nVote".EB."restEB"."allEB".ThR"maxT_hR".Tumor."restT"."allT".txt
Here is an example result file:
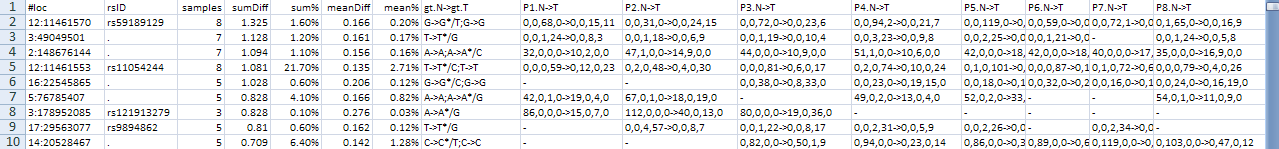
It has 8 columns plus one column for each patient:
loc chr:position
rsID rs# or '.'
samples No. of samples supporting this site as somatic mutated
sumDiff sum of differences between empirical Bayes adjusted
tumor and normal variation rates
sum% sum of percentage of top positions of supporting samples
meanDiff mean of differences between empirical Bayes adjusted
tumor and normal variation rates, = sumDiff/samples
mean% sum of percentage of top positions of supporting samples,
= meanDiff/samples
gt.N->gt.T genotype of normal -> genotype of tumor, could be multiple
pairs and sperated with ';'
P1.N->T patient 1: a,c,g,t of Normal -> a,c,g,t of Tumor; counts are
after MQ and BQ filtering; if the sample doesn't support the
site as somatic mutated, then '-'
... additional patients
In addition, there are three intermediate files for each patient. Their file names follow the patterns:
- "smN"."typeN"."smT"."typeT".ToAnalyzeBaseSub.txt.EB."restEB"."allEB".ThR"maxT_hR".inputEB
- "smN"."typeN"."smT"."typeT".ToAnalyzeBaseSub.txt.EB."restEB"."allEB".ThR"maxT_hR".R_re
- "smN"."typeN"."smT"."typeT".ToAnalyzeBaseSub.txt.EB."restEB"."allEB".ThR"maxT_hR".Tumor."restT"."allT".txt
IV. Running SMUG_LOH.region.pl
The script SMUG_LOH.region.pl does the following:- Read in a SMUG_LOH output file.
- Identify LOH regions.
Arguments for SMUG_LOH.region.pl
-f a SMUG_LOH output file (*.ToAnalyzeLOH.txt)
-minLOH minimum LOH score for LOH region detection, default = 30
-minSNPs minimum number of continuous SNPs required to define a
LOH region, default = 2
Example command to run SMUG_LOH.region.pl
perl SMUG_LOH.region.pl -f 1055QC0011.Normal.1055QC0031.Tumor.ToAnalyzeLOH.txt \ -minLOH 30.0 \ -minSNPs 2
Output files of SMUG_LOH.region.pl
There is one output file for each patient, with file name following the pattern:
- "smN"."tpeN"."smT"."typeT".ToAnalyzeLOH.txt.LOH."minLOH".SNPs."minSNPs".txt
Here is an example result file:

It has 3 columns:
region chr:startPosition-endPosition nSNPs Number of contiguous sites supporting the region regionLength length of region in bp
Ideas, requests, problems regarding Vanderbilt Biostatistics Wiki? Send feedback