Advanced Sequence Automated Pipeline (ASAP)
URL of this page: http://biostat.mc.vanderbilt.edu/ASAP Program: asap-v1.1.7.tgz Paper: ASAP an environment for automated preprocessing of sequencing data Torstenson E, Li B, Li C (2012) User Manual: ASAP_Reference_Guide-v1.1.7-1.pdf Tutorial: asap_example-1.1.4.tgz Tutorial should take about 15 minutes on modern systems Known Issues, bugs and future enhancement considerations are here Change list: ChangesIntroduction
 As next-generation sequencing becomes more affordable, researchers who wish to process the data themselves are finding the task to be both time consuming and error prone. The processing itself requires the use of many different programs, each of which has it’s own set of parameters and usage requirements. Some of these programs are under rapid development and change drastically after only a few months time. As a result, processing this data requires much more than processor time. End users must configure each command executed for their particular set of data, track successes and rerun failures, ensure the integrity of their data and maintain records of which settings were used for use when writing up their results.
As next-generation sequencing becomes more affordable, researchers who wish to process the data themselves are finding the task to be both time consuming and error prone. The processing itself requires the use of many different programs, each of which has it’s own set of parameters and usage requirements. Some of these programs are under rapid development and change drastically after only a few months time. As a result, processing this data requires much more than processor time. End users must configure each command executed for their particular set of data, track successes and rerun failures, ensure the integrity of their data and maintain records of which settings were used for use when writing up their results.
ASAP was designed to alleviate these issues by providing a modular system to allow users with different needs to process their data with a minimal amount of effort. In addition to minimizing human involvement, ASAP is designed to work on the researcher’s local computer cluster, if one is available.
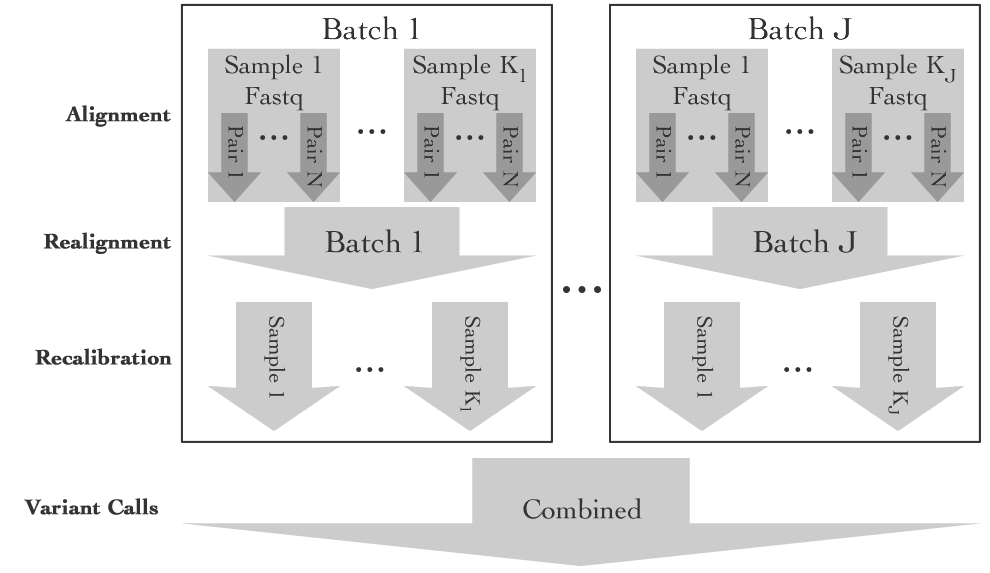
The program breaks the processing into 4 steps: Alignment, Realignment, Recalibration and Variant Calling. Processing is performed by tools produced by the research community itself, include bwa, samtools, GATK and a few others. Each of the four major steps might have subtle variations, such as how the samples are grouped during processing. Some steps, such as calling variants, even allow users to decide to use different underlying programs, such as using either samtools or GATK to call their SNPs. With the exception of Alignment, all steps are optional. So, researchers can process their data with or without realignment and recalibration, depending on their needs. Researchers are also able to modify the behavior of the underlying applications by changing the parameters that are passed at each step. As a result, ASAP offers a highly flexible mechanism for processing sequence data. As an added benefit, all configuration settings that determine how processing is done can be saved to a single configuration file, which can be shared with other users, or reused for processing other data.
While ASAP was designed for processing human fastq data into bam and vcf files, it should be suitable for use with any paired end reads as long as the user can obtain a reference genome and can be used by the various tools currently in use by the program.
In addition to processing data from unaligned through to variant calls, with the addition of import, ASAP can also be used as a simple way to call variants on very large datasets as data is in batches over time. ASAP will execute only those steps necessary.
Installation
To install, simply download the file and uncompress it: tar zxvf asap_v1.0.0.tgz You can then move the resulting directory to wherever you like. Once it has been moved into it's new home, it is strongly recommended that users add the directory to their PATH.A Note on GATK 2.x
With the release of GATK 2.x, Broad Institute began requiring a login for downloads of the complete version of GATK in order to license the use of the software differently for educational and non-educational groups. As such, ASAP is unable to download and install GATK 2.x versions for the user. Because of this, the default setting for ASAP is to use GATK 1.6.x, but users wish to download a newer version of GATK themselves may take advantage of this version by calling ASAP in a slightly different way. Please see the manual or the tutorial for more details on how to do this. Please note, that ASAP requires the full version of GATK 2.x as opposed to the lite version.Contact
Please send your comments and suggestions to asap-users@googlegroups.com (membership required)Ideas, requests, problems regarding Vanderbilt Biostatistics Wiki? Send feedback